Our struggle with HIV/AIDS epitomizes societies' millennia-old fight with microbial pathogens. One goal of HIV research is to generate effective interventions that will allow us to: 1) prevent further spread of HIV (vaccines and behavioural changes), and 2) eliminate the virus from those already infected (antivirals).
This sterilizing immunity - as it is referred to - has been a long sought-after goal for a number of viruses, yet widespread use of highly-active
anti-retroviral therapy (HAART) fails to completely remove the virus from an individual patient. Somewhere, somehow HIV is continuously replicating in your body. But how and why is this possible?
 |
| The spread of HIV from Dendritic cells to T lymphocytes or T to T cells may allow escape from antivirals. http://pathmicro.med.sc.edu |
One potential mechanism is that
HIV lies in a latent state - anatomical (central nervous system) or biochemical (following DNA integration and before gene expression) - where these drugs cannot inactivate it.
But researchers, headed by
David Baltimore at the California Institute of Technology, have come forward with both theoretical and experimental evidence suggesting a novel mechanism that explains how the virus may be able to circumvent HAART treatment through continuous replication and direct cell-cell spread. Their results were published in Nature last week. See
here.
What is cell-cell spread?
Viruses can infect new cells via a number of mechanisms. The most well-characteristic being via cell-free virus particles (see computer simulation paper
here). In this, new viruses are released from the originally infected cell and diffuse to an uninfected one nearby, thus establishing a novel infection.The problem with this is that it is pretty inefficient. For one, the viruses could be carried anywhere and even if they reach a cell, it may not be the right one. One other, more efficient means of transmission is through direct cell-cell spread, which generally takes diffusion out of the picture.
 |
| Model of how HIV moves from cell-cell. A) Dendritic cell (DC) with natural fold-like projections. B) DC picks up virus particles in red C) virus is held in vesicles within cell, D) T cell projections induced, E) formation of the virological synapse, F) release, binding and entry of HIV into T cells. (Felts, et al 2010) |
HIV and its close relative, the human T lymphotropic virus (HTLV-1) have been shown to move from one cell to another through this mechanism.
Original paper
here
Initial visualisation
In-depth 3D structural analysis
Check out the great videos
here
Following interaction with dendritic cells or T cells, the virus directs the assembly of a structure (referred to as the virological synapse) linking the two cells via alterations of the cytoskeleton which causes the two plasma membranes to come close together. This structure initially derives from the target cell.
It is here where new virus particles are released into the gap that has formed, thereby increasing the efficiency of spread by directing transmission and limiting the effects of diffusion.This synaptic structure, ultimately acts to concentrate both virus and receptors at defined subcellular locations. This kind of spread may allow the virus to become essentially invisible to our bodies' immune defences, such as neutralizing antibodies, and even antiviral drugs.
How does this allow escape from treatment?
 |
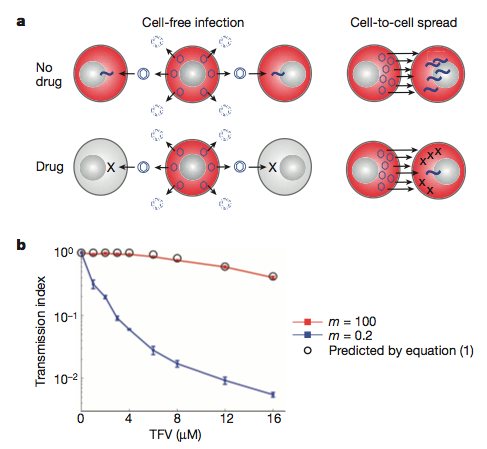
| Their model: a) inefficient cell-free infection will be eradicated with antvirals. The more efficient direct cell-cell spread will persist as it involves many more virus particles. b) The mathematical model (backed up by experimental data) showing the loss of infection (transmission index) with increasing concentration of antiviral (TFV) when we have few (m =0.2) or many (m = 100) virus particles. |
 |
| Expanding the model |
Initially, they generated - and experimentally verified - a mathematical model of how HIV transmission may differ during antiretroviral treatment under cell-free (inefficient - low virus transmission) or direct cell-cell spread (efficient - high virus transmission) mechanisms. Their results indicate that when there are many viruses around, the infection is more resistant to the drugs. This, they suggest, is due to the increased probability that at least one virus particle will not interact with the antiviral.
This observation extended to direct cell-cell spread when they introduced previously infected cells with non-infected ones and measured transmission of virus. Both the experimental and theoretical modelling suggest that this spread could be responsible for the ongoing replication seen in patients being treated with antivirals.
What does this mean for HIV therapy? Well, this work brings experimental evidence on the already known process of ongoing HIV replication in the face of antivirals. Through experimentally identifying this mechanism, the researchers here have uncovered a weak-point in HIV biology, one that is not currently being targeted. This method of cell-cell spread may also facilitate escape from neutralizing antibody responses through physically preventing their interaction with HIV proteins. How then might our own bodies combat this virus if its ability to more from cell-cell was inhibited? This work further adds to the growing body of work highlighting direct cell-cell spread as a principle mode of transmission for these retroviruses.
 Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, & Baltimore D (2011). Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature PMID: 21849975
Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, & Baltimore D (2011). Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature PMID: 21849975
















































{kind=link}